As well as telling us how many bacterial species make up a microbial community, 16S rRNA gene analysis can give us an idea of the abundance of each species. It would be great if things were simple. Let's say I analyse 100 sequences from an environmental sample. My analysis shows that 50 sequences are from the phylum Firmicutes, 30 are Proteobacteria and 20 belong to Bacteroidetes. So logically, I can assume that of all the bacteria in the sample 50% are Firmicutes, 30% are Proteobacteria and 20% are Bacteroidetes. However, if I've used PCR to amplify the DNA in my sample, I have to assume that all of the genes are amplified equally. This assumption is wrong. Some sequences will be easier to copy using PCR than others. This discrepancy is called differential or preferential amplification. Differential amplification is a bias introduced by PCR
which cannot always be corrected. Several factors have been
identified as causing differential amplification of rDNA.
-
The rRNA gene copy number (rrn operon number) and genome size differ between species.Bacteria can have between 1 and 10 copies of the rRNA gene within their genome (1). What’s more, the copy number of rRNA genes doesn’t necessarily correspond to a regular increase in PCR product so even if we knew the rrn operon number of each species, we couldn't correct for it. Other factors such as density of rRNA genes and the percentage of the genome composed of rRNA genes have also been theorised to affect the efficiency of PCR amplification (2,3). There are online databases that have information on the rrn operon number of different bacterial species. For example, here we can see that Lactobacillus acidophilus has 4 16S rRNA gene copies in its genome, according to two different studies.
-
Not all rRNA genes from the same species have exactly the same sequence.By reviewing pairs of sequences from the same species in databases of rRNA gene sequences, it has been estimated that up to 48% of sequence pairs have more variation than would be expected from sequencing errors. This variation is different between taxa, so there is no easy mathematical correction for this observation (4). Another study of differences in sequence found that 16S rRNA gene sequences from strains of Paenicabillus polymyxa differed from each other by one to eight nucleotides at ten places in the V6 to V8 regions (5). Intraspecific heterogeneity (differences within the same species) can complicate the quantification of bacteria and lead to an overestimation of diversity (1, 2).
-
Differences in G+C content between sequencesThe G-C content of a DNA sequence is the proportion of base pairs that are G-C instead of A-T. The G-C content is important as it defines how stable a DNA molecule will be at higher temperatures. This is the central tenant on which temperature and denaturing gradient gel electrophoresis separates DNA molecules based on their sequences. Basically, DNA molecules with a higher G-C content are more thermostable than those with a low G-C content. This is because of the stacking of the base pairs, which is beyond the scope of this article, but keen biochemists can read up about it here. rDNA sequences that have a lower G+C content denature in the PCR and so may be preferentially amplified. This effect can be reduced by adding 5% acetamide which also stops primers from binding preferentially to different DNA strands (6).


The top strand has a lower G-C content than the bottom one and so will denature more readily. -
Sequences outside the rRNA gene can inhibit amplification.Other DNA sequences and secondary structural features of the bacterial genome that serves as the original template can inhibit PCR amplification of the rRNA gene. DNA isn't a straight molecule. It curls up on itself and has other proteins bound to it. These secondary structural features can physically get in the way of primer binding. The inhibitory effect of these secondary structures varies depending on which variable section is targeted by the primers (7–9). One group found they couldn't overcome the inhibitory effect of by using DNA denaturing cosolvents such as DMSO and glycerol or other techniques such as touchdown PCR. Instead, they suggested that the effect can be minimised by using at least two primer sets targeting different variable sections of the rRNA gene in separate PCRs, then comparing the results (7).
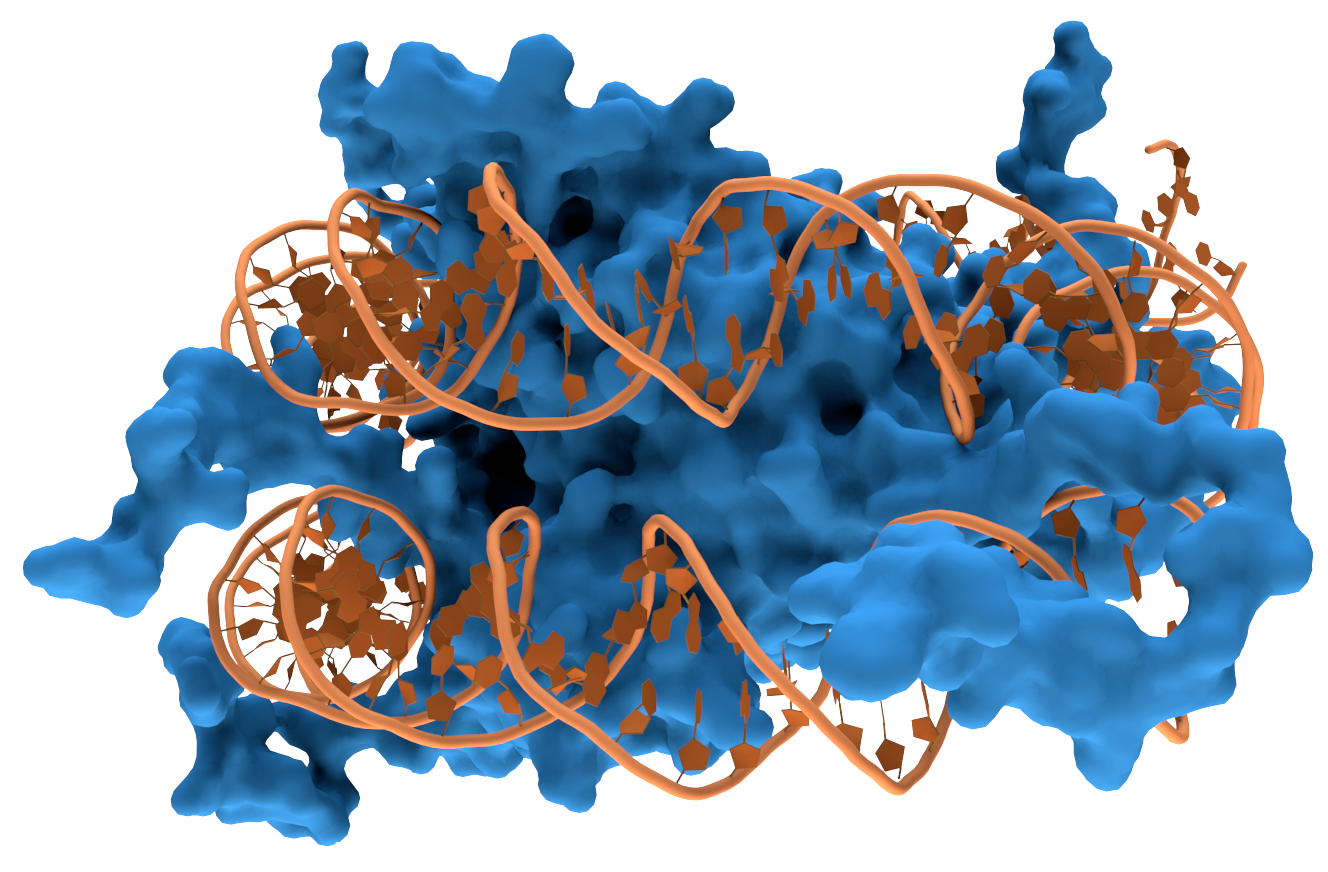
A strand of DNA wrapped around a DNA binding protein which could obstruct PCR amplification
Thomas Splettstoesser, Nucleosome1, CC BY-SA 3.0


{kind=link}
-
Increasing template concentration reduces the rate of amplification.In the typical description of PCR, the DNA strands denature and the primer binds. But what's stopping the DNA strands from just reannealing to each other instead of a primer once the temperature drops? The answer is... nothing, except that usually the other DNA strand has floated away a bit and the nearest thing to bind to is a primer. However, a critical concentration of template DNA exists at which reannealing of DNA strands is favoured over primer binding. When the concentration of template DNA reaches and goes over this critical concentration, amplification is reduced. This allows other rDNA templates to be more effectively amplified in subsequent PCR cycles and will alter the relative abundance of rDNA sequences within the sample. This amplification bias is less likely to occur in samples with a wide variety of rDNA sequences at relatively low concentrations (9).
-
Specificity of primers to the template DNA.Even if universal primers are used, there is evidence to suggest that there is differential binding between primers and template DNA from different bacterial species. Even single mismatches between primers and template DNA can reduce binding (10). Suboptimal binding will result in decreased amplification of the respective template compared to others (11). While lowering the annealing temperature will allow for mismatches, it can increase non-specific primer binding and unwanted products (12).
-
DNA contamination of PCR.Introduction of DNA to the sample can occur either through unintentional transfer of DNA from previous amplifications (tube-to-tube contamination) or by contamination of PCR reagents (11). This is a particular problem for reagents such as DNA polymerase whose manufacture involves the use of Escherichia coli (1). To protect against this, a negative control must always be included which is handled the same as other samples, except that no template DNA is added. Reagents should also be pre-treated with UV light or uracil DNA glycosylase to remove contaminating DNA (13).
Let's have a look at this paper investigating poultry intestinal bacteria using denaturing gel gradient electrophoresis. They have this to say about their PCR:
"Primers7 (50 pmol of each per reaction mixture; primer 2, 5′-ATTACCGCGGCTGCTGG-3′, and primer 3 with a 40-base G-C clamp (Sheffield et al., 1989; Muyzer et al., 1993), 5′-CGCCCGCCGCGCGCGGCGGGCGGGG CGGGGGCACGGGGGGCCTACGGGAGGCAGCAG- 3′) were mixed with Jump Start Red-Taq Ready Mix,5 according to the kit instructions, 250 ng of pooled (50 ng/ chicken) template DNA from five chickens in each group, and 5% (wt/vol) acetamide to eliminate preferential annealing (Reysenbach et al., 1992). Amplifications were on a PTC-200 Peltier Thermal Cycler8 with the following program: 1) denaturation at 94.9°C for 2 min; 2) subsequent denaturation at 94.0°C for 1 min; 3) annealing at 67.0°C for 45 s, −0.5°C per cycle [touchdown to minimise spurious by-products (Don, 1991; Wawer and Muyzer, 1995)]; 4) extension at 72.0°C for 2 min; 5) repeat steps 2 to 4 for 17 cycles; 6) denaturation at 94°C for 1 min; 7) annealing at 58.0°C for 45 s; 8) repeat steps 6 to 7 for 12 cycles; 9) extension at 72.0°C for 7 min; 10) 4.0°C final."
Although they've taken precautions (highlighted in bold) to minimise certain factors that contribute to differential amplification it's impossible to correct for others, such as a different rrn operon number or intraspecific heterogeneity. In light of this, any experiment using PCR will introduce some biases and won't produce a 100% accurate picture of the microbial community being studied.
Although PCR is an imperfect technique, it is currently the only reliable way of amplifying DNA from environmental samples. After amplification, the DNA from a sample can either be analysed directly using fingerprinting techniques such as DGGE, TGGE and T-RFLP or individual DNA fragments can be sequenced to identify the bacteria present and build phylogenetic trees. While modern sequencing platforms like Illumina and 454 pyrosequencing require no additional steps after PCR, older studies which relied on chain-termination sequencing had to build clone libraries of sampled DNA. The creation of a clone library is a lengthy process and can also introduce biases which affect results.
References
1. Osborn M A, Smith CJ. Molecular Microbial Ecology. Vol. 51. 2009. 370 p.
2. Stackebrandt E, Pukall R, Ulrichs G, Rheims H. Analysis of 16S rDNA clone libraries: part of the big picture. Proc 8th Int Symp Microb Ecol Microb Biosyst new Front Atl Canada Soc Microb Ecol Halifax, Nov Scotia, Canada
"Primers7 (50 pmol of each per reaction mixture; primer 2, 5′-ATTACCGCGGCTGCTGG-3′, and primer 3 with a 40-base G-C clamp (Sheffield et al., 1989; Muyzer et al., 1993), 5′-CGCCCGCCGCGCGCGGCGGGCGGGG CGGGGGCACGGGGGGCCTACGGGAGGCAGCAG- 3′) were mixed with Jump Start Red-Taq Ready Mix,5 according to the kit instructions, 250 ng of pooled (50 ng/ chicken) template DNA from five chickens in each group, and 5% (wt/vol) acetamide to eliminate preferential annealing (Reysenbach et al., 1992). Amplifications were on a PTC-200 Peltier Thermal Cycler8 with the following program: 1) denaturation at 94.9°C for 2 min; 2) subsequent denaturation at 94.0°C for 1 min; 3) annealing at 67.0°C for 45 s, −0.5°C per cycle [touchdown to minimise spurious by-products (Don, 1991; Wawer and Muyzer, 1995)]; 4) extension at 72.0°C for 2 min; 5) repeat steps 2 to 4 for 17 cycles; 6) denaturation at 94°C for 1 min; 7) annealing at 58.0°C for 45 s; 8) repeat steps 6 to 7 for 12 cycles; 9) extension at 72.0°C for 7 min; 10) 4.0°C final."
Although they've taken precautions (highlighted in bold) to minimise certain factors that contribute to differential amplification it's impossible to correct for others, such as a different rrn operon number or intraspecific heterogeneity. In light of this, any experiment using PCR will introduce some biases and won't produce a 100% accurate picture of the microbial community being studied.
Although PCR is an imperfect technique, it is currently the only reliable way of amplifying DNA from environmental samples. After amplification, the DNA from a sample can either be analysed directly using fingerprinting techniques such as DGGE, TGGE and T-RFLP or individual DNA fragments can be sequenced to identify the bacteria present and build phylogenetic trees. While modern sequencing platforms like Illumina and 454 pyrosequencing require no additional steps after PCR, older studies which relied on chain-termination sequencing had to build clone libraries of sampled DNA. The creation of a clone library is a lengthy process and can also introduce biases which affect results.
References
1. Osborn M A, Smith CJ. Molecular Microbial Ecology. Vol. 51. 2009. 370 p.
2. Stackebrandt E, Pukall R, Ulrichs G, Rheims H. Analysis of 16S rDNA clone libraries: part of the big picture. Proc 8th Int Symp Microb Ecol Microb Biosyst new Front Atl Canada Soc Microb Ecol Halifax, Nov Scotia, Canada
3. Farrelly V, Rainey F a, Stackebrandt E, Farrelly V, Rainey F a. Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species . These include : Effect of Genome Size and rrn Gene Copy Number on PCR Amplification of 16S rRNA Genes from a Mixture of Bacterial S. 1995;61(7):2798–801.
4. Clayton RA, Sutton G, Hinkle Jr. PS, Bult C, Fields C. Intraspecific variation in small-subunit rRNA sequences in GenBank: why single sequences may not adequately represent prokaryotic taxa. Int. J. Syst. Bacteriol. 1995;45:595–9.
5. Nubel U, Engelen B, Felske A, Snaidr J, Wieshuber A, Amann RI, et al. Sequence Heterogeneities of Genes Encoding 16S rRNAs in Paenibacillus polymyxa Detected by Temperature Gradient Gel Electrophoresis. J Bacteriol. 1996;178(19):5636–43.
6. Reysenbach AL, Giver LJ, Wickham GS, Pace NR. Differential amplification of ribosomal RNA genes by polymerase chain reaction. Appl Env Microbiol [Internet]. 1992;58(10):3417–8.
7. Hansen MC, Tolker-Nielsen T, Givskov M, Molin S. Biased 16S rDNA PCR amplification caused by interference from DNA flanking the template region. FEMS Microbiol Ecol. 1998;26(2):141–9.
8. Rainey F. A., Ward N, Sly L. I., Stackebrandt E. Dependence on the taxon composition of clone libraries for PCR amplified, naturally occurring 16S rDNA, on the primer pair and the cloning system used. Experientia. 1994;50(9):796–7.
9. Suzuki MT, Giovannoni SJ. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. 1996;62(2):2–8.
10. Dahllöf I. Molecular community analysis of microbial diversity. Curr Opin Biotechnol. 2002;13(3):213–7.
11. Wintzingerode F, Göbel UB, Stackebrandt E. Determination of microbial diversity in environmental samples: pitfalls of PCR-based analysis. FEMS Microbiol Rev. 1997;21:213–29.
12. Ishii K, Fukui M. Optimization of Annealing Temperature to Reduce Bias Caused by a Primer Mismatch in Multitemplate PCR. Appl Environ Microbiol. 2001;67(8):3753–5.
13. Niederhauser C, Höfelein C, Wegmüller B, Lüthy J, Candrian U. Reliability of PCR decontamination systems. Genome Res. 1993;4(2):117–23.
No comments:
Post a Comment